Nextflow Workflow
Nextflow Workflow
mkdir workflow
cd workflow
Our previous episodes have shown us how to parameterize workflows using params, move data around a workflow using channels, and define individual tasks using processes. In this episode, we will cover how to connect multiple processes to create a workflow.
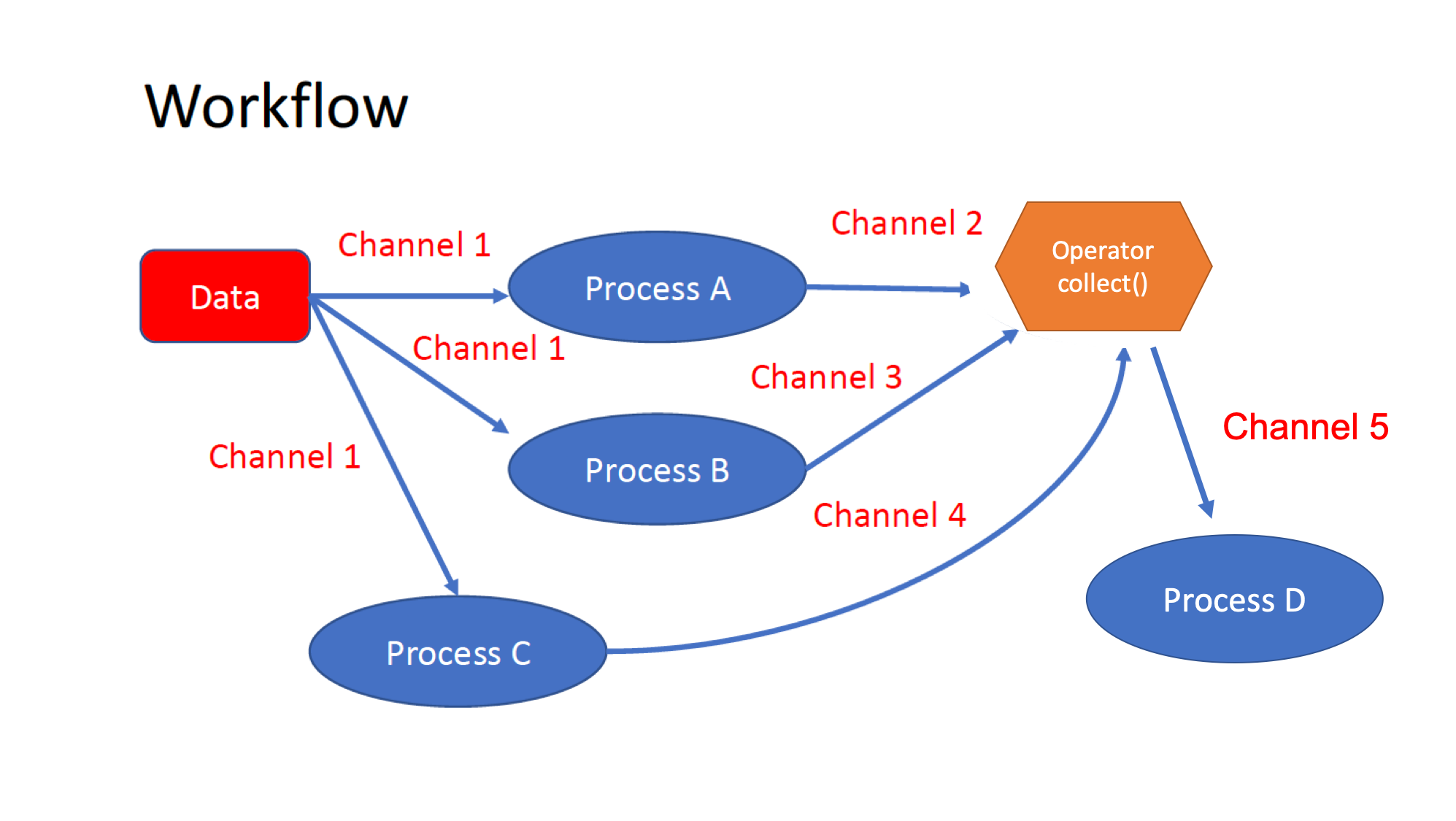
-
The workflows can be represented as graphs where the nodes are the
processesand the edges are thechannels. The processes are blocks of code that can be executed - such as scripts or programs - while the channels are asynchronous queues able to connect processes among them via input/output. Some methods, calledoperators, are provided for reshaping and combining the channels. -
Processes are independent of each other and can be run in parallel, depending on the number of elements in a channel. In the previous example, processes A, B, and C can be run in parallel, and only when they ALL end, the process D is triggered. An operator is used for gathering together all the elements generated by the channels 2, 3, and 4.
Workflow definition
- We can connect processes to create our pipeline inside a
workflowscope. The workflow scope starts with the keywordworkflow, followed by an optional name and finally the workflow body delimited by curly brackets{}.
Implicit workflow - A workflow definition that does not declare any name is assumed to be the main workflow, and it is implicitly executed. Therefore, it’s the entry point of the workflow application.
- To combine multiple processes, invoke them in the order they would appear in a workflow.
-
When invoking a process with multiple inputs, provide them in the same order in which they are declared in the
inputblock of the process. - A workflow component can access any variable and parameter defined in the outer scope. In the example below, the
params.genomeandparams.readscan be accessed inside theworkflowscope:
Create a new workflow.nf; add the following code block and run nextflow run workflow.nf:
//workflow.nf
nextflow.enable.dsl=2
// Initialize required parameters
params.outdir = 'results'
params.genome = "/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta"
params.reads = "/workspace/nextflow_tutorial/data/trimmed_fastq/SRR2584863_{1,2}.trim.fastq.gz"
workflow {
// Create channel from path for Reference Genome
ref_ch = Channel.fromPath( params.genome, checkIfExists: true )
// Create channel from file-pairs for Input Fastq files
reads_ch = Channel.fromFilePairs( params.reads, checkIfExists: true )
//index process takes 1 input channel as an argument
BWA_INDEX( ref_ch )
//bwa align process takes 2 input channels as arguments
BWA_ALIGN( BWA_INDEX.out, reads_ch )
}
process BWA_INDEX {
tag {"BWA_INDEX ${genome}"}
label 'process_low'
publishDir "${params.outdir}/bwa_index", mode: 'copy'
input:
path( genome )
output:
tuple path( genome ), path( "*" )
script:
"""
bwa index ${genome}
"""
}
/*
* Align reads to reference genome & create BAM file.
*/
process BWA_ALIGN {
tag {"BWA_ALIGN ${sample_id}"}
label 'process_medium'
publishDir "${params.outdir}/bwa_align", mode: 'copy'
input:
tuple path( genome ), path( "*" )
tuple val( sample_id ), path( reads )
output:
tuple val( sample_id ), path( "${sample_id}.aligned.bam" )
script:
"""
INDEX=`find -L ./ -name "*.amb" | sed 's/.amb//'`
bwa mem \$INDEX ${reads} > ${sample_id}.aligned.sam
samtools view -S -b ${sample_id}.aligned.sam > ${sample_id}.aligned.bam
"""
}
-
In this example, the
BWA_INDEXprocess is invoked first, and theBWA_ALIGNprocess second. -
A process output can also be accessed using the
outattribute for the respectiveprocess object. -
The
BWA_INDEX.outobject is passed as the first argument to theBWA_ALIGNprocess, and thereads_chchannel is passed as the second argument (in the same order they are declared as inputs in the process). -
When a process defines two or more output channels, each of them can be accessed using the list element operator, e.g. out[0], out[1], or using named outputs.
Process Named Output
-
The process
outputdefinition allows the use of theemit:option to define a named identifier that can be used to reference the channel in the external scope. -
For example, in the script below, we name the output from the
BWA_INDEXprocess asbwa_indexusing theemit:option. We can then reference the output asBWA_INDEX.out.bwa_indexin the workflow scope.
Modify workflow.nf as shown below and run nextflow run workflow.nf:
//workflow.nf
nextflow.enable.dsl=2
// Initialize required parameters
params.outdir = 'results'
params.genome = "/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta"
params.reads = "/workspace/nextflow_tutorial/data/trimmed_fastq/SRR2584863_{1,2}.trim.fastq.gz"
workflow {
// Create channel from path for Reference Genome
ref_ch = Channel.fromPath( params.genome, checkIfExists: true )
// Create channel from file-pairs for Input Fastq files
reads_ch = Channel.fromFilePairs( params.reads, checkIfExists: true )
//index process takes 1 input channel as an argument
BWA_INDEX( ref_ch )
//bwa align process takes 2 input channels as arguments
BWA_ALIGN( BWA_INDEX.out.bwa_index, reads_ch )
}
process BWA_INDEX {
tag {"BWA_INDEX ${genome}"}
label 'process_low'
publishDir "${params.outdir}/bwa_index", mode: 'copy'
input:
path( genome )
output:
tuple path( genome ), path( "*" ), emit: bwa_index
script:
"""
bwa index ${genome}
"""
}
/*
* Align reads to reference genome & create BAM file.
*/
process BWA_ALIGN {
tag {"BWA_ALIGN ${sample_id}"}
label 'process_medium'
publishDir "${params.outdir}/bwa_align", mode: 'copy'
input:
tuple path( genome ), path( "*" )
tuple val( sample_id ), path( reads )
output:
tuple val( sample_id ), path( "${sample_id}.aligned.bam" ), emit: aligned_bam
script:
"""
INDEX=`find -L ./ -name "*.amb" | sed 's/.amb//'`
bwa mem \$INDEX ${reads} > ${sample_id}.aligned.sam
samtools view -S -b ${sample_id}.aligned.sam > ${sample_id}.aligned.bam
"""
}
Quick Recap
- A Nextflow workflow is defined by invoking
processesinside theworkflowscope.- A process is invoked like a function inside the
workflowscope, passing any required input parameters as arguments. e.g.,BWA_INDEX( ref_ch ).- Process outputs can be accessed using the
outattribute for the respectiveprocess.- Multiple outputs from a single process can be accessed using the
[]or output name.