Nextflow Processes
Nextflow Processes
We now know how to create and use Channels to send data around a workflow. We will now see how to run tasks within a workflow using processes.
Processes
- A
processis the way Nextflow executes commands you would run on the command line or custom scripts. - Processes can be thought of as particular tasks or steps in a workflow, e.g. an alignment step (
bwa mem) in variant-calling analysis. - Processes are independent of each other (don’t require another process to execute) and cannot communicate/write to each other.
- It is the channels that pass the data from one process to another, and we do this by having the processes define
inputandoutputchannels.
For example, below is the command line you would run on a terminal to create an index for the ecoli genome to be used with the BWA aligner:
bwa index data/ref_genome/ecoli_rel606.fasta
- In Nextflow, the process definition starts with the keyword
process, followed by the process name and finally the process body delimited by curly brackets{}. - The process body must contain a string which represents the command or, more generally, a script that is executed by it.
process BWA_INDEX {
script:
"""
bwa index data/ref_genome/ecoli_rel606.fasta
"""
}
- In order to run the process, we need to add it to a
workflowblock below the process. - The workflow scope starts with the keyword
workflow, followed by an optional name and finally the workflow body delimited by curly brackets{}. We will learn more about the workflow block in detail in the next chapter.
Create a new process_index.nf file; add the following code block and nextflow run process_index.nf:
//process_index.nf
nextflow.enable.dsl=2
process BWA_INDEX {
script:
"""
bwa index "/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta"
"""
}
workflow {
BWA_INDEX()
}
Output:
N E X T F L O W ~ version 21.04.3
Launching `process_index.nf` [cheesy_coulomb] - revision: 72a7a10fb7
executor > local (1)
[00/fcc341] process > BWA_I... [100%] 1 of 1 ✔
Definition blocks
- The previous example was a simple process with no defined inputs and outputs that ran only once.
- To control inputs, outputs, and how a command is executed, a process may contain five definition blocks:
directives: allow the definition of optional settings that affect the execution of the current process, e.g., the number of CPUs a task uses and the amount of memory allocated.inputs: Define the input dependencies, usually channels, which determine the number of times a process is executed.outputs: Defines the output channels used by the process to send results/data produced by the process.whenclause: Allows you to define a condition that must be verified in order to execute the process.- The
scriptblock: A statement within quotes that defines the commands that are executed by the process to carry out its task.
The syntax is defined as follows:
process < NAME > {
[ directives ]
input:
< process inputs >
output:
< process outputs >
when:
< condition >
[script|shell|exec]:
< user script to be executed >
}
In the next steps, we will learn how to use these definition blocks to create more complex processes with input and output channels.
Let’s create a new process that takes a reference genome as input and creates a BWA index for it. First, create a new file called process_index_with_input.nf.
// process_index_with_input.nf
nextflow.enable.dsl=2
process BWA_INDEX {
input:
path ref_genome
output:
path "${ref_genome}.bwt", emit: index
script:
"""
bwa index "$ref_genome"
"""
}
workflow {
ref_genome_ch = Channel.fromPath("/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta")
BWA_INDEX(ref_genome_ch)
}
In this example, we have defined an input channel ref_genome that takes a reference genome file as input. The output channel index emits the BWA index file. The process will now execute the bwa index command using the input reference genome file.
To run the new process, execute nextflow run process_index_with_input.nf. You should see the output similar to the previous example, but this time the process has taken input from the ref_genome_ch channel and produced an output.
As you start creating more complex workflows, you can use the different definition blocks within processes to control their inputs, outputs, and execution behavior. This allows you to build modular, reusable, and efficient pipelines in Nextflow.
Script
- At minimum, a process block must contain a
scriptblock. - The
scriptblock is a String “statement” that defines the command that is executed by the process to carry out its task. These are normally the commands you would run on a terminal. - A process contains only one
scriptblock, and it must be the last statement when the process containsinputandoutputdeclarations.
Example we have seen before:
// process_index.nf
nextflow.enable.dsl=2
process BWA_INDEX {
script:
"""
bwa index "/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta"
"""
}
workflow {
BWA_INDEX()
}
- By default, the process command is interpreted as a Bash script.
- However, any other scripting language can be used by simply starting the script with the corresponding Shebang declaration.
For example, if you wanted to use a Python script in a process, you could include the Python shebang (#!/usr/bin/env python) at the beginning of the script block:
// process_with_python.nf
nextflow.enable.dsl=2
process PYTHON_SCRIPT {
debug true // enable debug mode which prints the stdout
script:
"""
#!/usr/bin/env python
import sys
print("Hello from Python!")
"""
}
workflow {
PYTHON_SCRIPT()
}
When you run this example with nextflow run process_with_python.nf, Nextflow will recognize the Python shebang and execute the script using the Python interpreter.
By allowing the use of different scripting languages, Nextflow provides flexibility in implementing custom tasks within your workflow.
Example - using python and R scripts
//process_python.nf
nextflow.enable.dsl=2
process PYSTUFF {
script:
"""
#!/usr/bin/env python
import gzip
reads = 0
bases = 0
with gzip.open('data/yeast/reads/ref1_1.fq.gz', 'rb') as read:
for id in read:
seq = next(read)
reads += 1
bases += len(seq.strip())
next(read)
next(read)
print("reads", reads)
print("bases", bases)
"""
}
workflow {
PYSTUFF()
}
//process_rscript.nf
nextflow.enable.dsl=2
process RSTUFF {
script:
"""
#!/usr/bin/env Rscript
library("ShortRead")
countFastq(dirPath="data/yeast/reads/ref1_1.fq.gz")
"""
}
workflow {
RSTUFF()
}
This allows the the use of a different programming languages which may better fit a particular job. However, for large chunks of code is suggested to save them into separate files and invoke them from the process script.
nextflow.enable.dsl=2
process PYSTUFF {
script:
"""
python myscript.py
"""
}
workflow {
PYSTUFF()
}
## Script parameters
- Similar to bash scripting, Nextflow uses the
$character to introduce variable substitutions. - The variable name to be expanded may be enclosed in braces
{variable_name}, which are optional but serve to protect the variable to be expanded from characters immediately following it which could be interpreted as part of the name. It is a good rule of thumb to always use the{}syntax. - A Nextflow variable can be used multiple times in the script block.
- In most cases, we do not want to hard code parameter values. A special Nextflow map variable
paramscan be used to assign values from the command line. You would do this by adding a key name to the params variable and specifying a value, likeparams.keyname = value
Modify process_index.nf as shown below and run with nextflow run process_index.nf:
//process_index.nf
nextflow.enable.dsl=2
params.genome = "/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta"
process BWA_INDEX {
script:
"""
bwa index ${params.genome}
"""
}
workflow {
BWA_INDEX()
}
Bash variables
-
Nextflow uses the same Bash syntax for variable substitutions,
$variable, in strings. However, Bash variables need to be escaped using the\character in front of\$variablename. -
In the example below, we will set the bash variable
REF_GENOMEto the value of$params.genome, and then useREF_GENOMEin our script block.
Modify process_index.nf as shown below and run with nextflow run process_index.nf:
//process_escape_bash.nf
nextflow.enable.dsl=2
params.genome = "/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta"
process BWA_INDEX {
script:
"""
REF_GENOME=${params.genome}
bwa index \$REF_GENOME
"""
}
workflow {
BWA_INDEX()
}
In this example, the Nextflow variable ${params.genome} is used to set the value of the Bash variable REF_GENOME, which is then used in the bwa index command. Note the use of the \ character to escape the $ when referencing the Bash variable (\$REF_GENOME).
Conditional script execution
- Sometimes you want to change how a process is run depending on some condition.
- In Nextflow scripts, you can use conditional statements such as the
ifstatement or any other expression evaluating to boolean valuetrueorfalse. - The
ifstatement uses the same syntax common to other programming languages such as Java, C, JavaScript, etc.
if( < boolean expression > ) {
// true branch
}
else if ( < boolean expression > ) {
// true branch
}
else {
// false branch
}
Example of a Nextflow script that uses if conditional:
//process_conditional.nf
nextflow.enable.dsl=2
params.aligner = 'kallisto'
params.transcriptome = "$projectDir/data/yeast/transcriptome/Saccharomyces_cerevisiae.R64-1-1.cdna.all.fa.gz"
params.kmer = 31
process INDEX {
script:
if( params.aligner == 'kallisto' ) {
"""
echo indexed using kallisto
kallisto index -i index -k $params.kmer $params.transcriptome
"""
}
else if( params.aligner == 'salmon' ) {
"""
echo indexed using salmon
salmon index -t $params.transcriptome -i index --kmer $params.kmer
"""
}
else {
"""
echo Unknown aligner $params.aligner
"""
}
}
workflow {
INDEX()
}
Inputs
- Processes are isolated from each other but can communicate by sending values and files via Nextflow channels into
inputandoutputblocks. - The
inputblock defines which channels the process is expecting to receive input from. - The number of elements in input channels determine the process dependencies and the number of times a process executes.
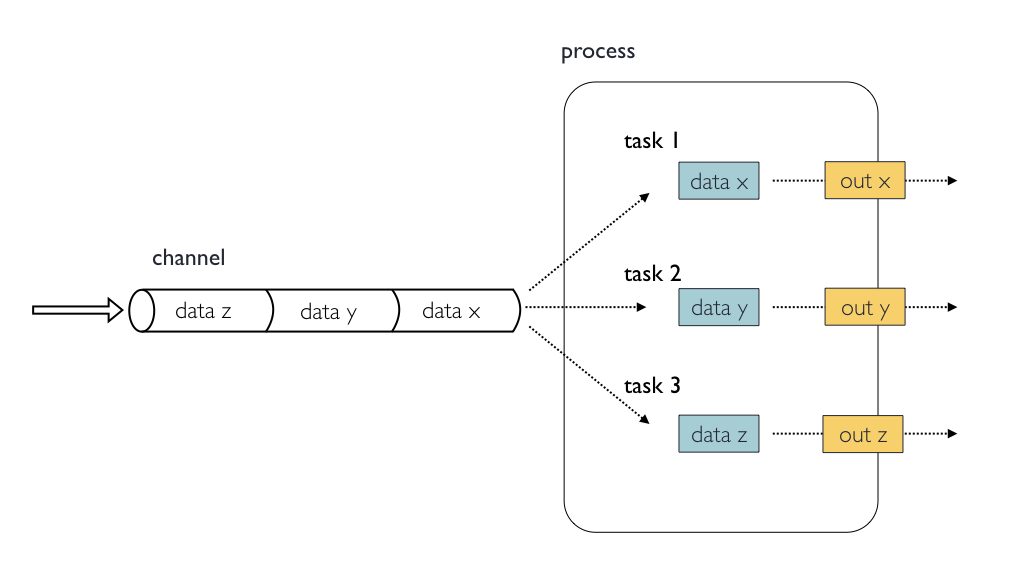
- You can only define one input block at a time and it must contain one or more input declarations. The input block follows the syntax shown below:
input:
<input qualifier> <input name>
- The input qualifier declares the type of data to be received. Types of input qualifiers:
val: Lets you access the received input value by its name in the process script.env: Lets you use the received value to set an environment variable named as the specified input name.path: Lets you handle the received value as a path, staging the file properly in the execution context.stdin: Lets you forward the received value to the process stdin special file.tuple: Lets you handle a group of input values having one of the above qualifiers.each: Lets you execute the process for each entry in the input collection.
Input values
- The
valqualifier allows you to receive value data as input. It can be accessed in the process script by using the specified input name, as shown in the following example:
Create a new process_input_value.nf file; add the following code and run nextflow run process_input_value.nf -process.echo:
//process_input_value.nf
nextflow.enable.dsl=2
process PRINTCHR {
input:
val chr
script:
"""
echo processing chromosome ${chr}
"""
}
chr_ch = Channel.of( 1..22,'X','Y' )
workflow {
PRINTCHR( chr_ch )
}
In the above example, we haven’t declared where the output of the
PRINTCHRprocess should go. If you execute withnextflow run process_input_value.nf, the script would run but you wouldn’t see any output printed on the screen. Hence, we add the argument-process.echoat run-time to display the standard output of process execution on screen. This won’t be necessary when we learn how to redirect output in the upcoming section. However, the argument-process.echocan be great for debugging.
Output:
[10/498dfc] process > PRINTCHR (24) [ 83%] 20 of 24 processing chromosome 3 processing chromosome 1 processing chromosome 2 ..truncated...In the above example, the process is executed 24 times; each time a value is received from the queue channel
chr_ch, it is used to run the process.
Channel order: The channel guarantees that items are delivered in the same order as they have been sent, but, since the process is executed in a parallel manner, there is no guarantee that they are processed in the same order as they are received.
Input files
- When you need to handle files as input, you should use the
pathqualifier. - Using the
pathqualifier means that Nextflow will stage the file in the process execution directory, and it can be accessed in the script by using the name specified in the input declaration. - The input file name can be defined dynamically by defining the input name as a Nextflow variable and referenced in the script using the
$variable_namesyntax. - For example, in the script below, we assign the variable name
genometo the input files using thepathqualifier. The file is referenced using the variable substitution syntax${genome}in the script block.
Modify process_index.nf as shown below and run nextflow run process_index.nf:
//process_input_file.nf
nextflow.enable.dsl=2
/*
* Index the reference genome for use by bwa and samtools.
*/
process BWA_INDEX {
input:
path genome
script:
"""
bwa index ${genome}
"""
}
ref_ch = Channel.fromPath("/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta")
workflow {
BWA_INDEX( ref_ch )
}
File Objects as inputs: When a process declares an input file, the corresponding channel elements must be file objects, i.e., created with the path helper function from the file-specific channel factories, e.g.,
Channel.fromPathorChannel.fromFilePairs.
Quick Recap:
- A Nextflow process is an independent step in a workflow.
- Processes can contain up to five definition blocks, including:
directives,inputs,outputs,whenclause, and finally, ascriptblock.- The
scriptblock contains the commands you would like to run.- A process should have a
script, but the other four blocks are optional.- Inputs are defined in the
inputblock with a type qualifier and a name.
Outputs
- We have seen how to input data into a process; now, we will see how to output files and values from a process.
- The
outputdeclaration block allows us to define the channels used by the process to send out the files and values produced. - An output block is not required, but if it is present, it can contain one or more output declarations.
The output block follows the syntax shown below:
output: <output qualifier> <output name>
Output values
- The type of output data is defined using output qualifiers.
- The
valqualifier allows us to output a value defined in the script. - If we want to capture a file instead of a value, we can use the
pathqualifier that can capture one or more files produced by the process over the specified channel. - Because Nextflow processes can only communicate through channels, if we want to share a value input into one process as input to another process, we would need to define that value in the output declaration block.
- When an output file name contains a
*or?character, it is interpreted as a pattern match. This allows capturing multiple files into a list and outputting them as a one-item channel. - Since all the files produced by the process are captured using
"*"in the output block, when the task is completed, all the output files are sent over the output channel. A downstreamoperatororprocessdeclaring the same channel asinputwill be able to receive it.
Modify process_index.nf as shown below and run nextflow run process_index.nf:
//process_output_value.nf
nextflow.enable.dsl=2
process BWA_INDEX {
input:
path genome
output:
path("*")
script:
"""
bwa index ${genome}
"""
}
ref_ch = Channel.fromPath("/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta")
workflow {
BWA_INDEX(ref_ch)
BWA_INDEX.out.view()
}
Output
N E X T F L O W ~ version 21.04.3 Launching `process_output_value.nf` [fervent_marconi] - revision: 1103ac760e executor > local (1) [9c/db0e50] process > BWA_INDEX (1) [100%] 1 of 1 ✔ [nextflow_tutorial/processes/work/9c/db0e50c168babb81cfb228bae0cb41/ecoli_rel606.fasta.amb, nextflow_tutorial/processes/work/9c/db0e50c168babb81cfb228bae0cb41/ecoli_rel606.fasta.ann, nextflow_tutorial/processes/work/9c/db0e50c168babb81cfb228bae0cb41/ecoli_rel606.fasta.bwt, nextflow_tutorial/processes/work/9c/db0e50c168babb81cfb228bae0cb41/ecoli_rel606.fasta.pac, nextflow_tutorial/processes/work/9c/db0e50c168babb81cfb228bae0cb41/ecoli_rel606.fasta.sa]
Note: Some caveats on glob pattern behavior:
- Input files are not included in the list of possible matches.
- Glob pattern matches against both files and directories path.
- When a two-stars pattern
**is used to recurse across directories, only file paths are matched, i.e., directories are not included in the result list.
Grouped inputs and outputs
-
So far, we have seen how to declare multiple input and output channels, but each channel was handling only one value at a time. However, Nextflow can handle groups of values using the
tuplequalifiers. -
In tuples, the first item is the grouping key, and the second item is the list of files.
[group_key,[file1,file2,...]]
- When using a channel containing a tuple, such as one created with the
.fromFilePairsfactory method, the corresponding input declaration must be declared with a tuple qualifier, followed by the definition of each item in the tuple.
Create a new file process_tuple_input.nf; add the following and run nextflow run process_tuple_input.nf -process.echo:
//process_tuple_input.nf
nextflow.enable.dsl=2
process TUPLEINPUT {
input:
tuple val(sample_id), path(reads)
script:
"""
echo ${sample_id}
echo ${reads}
"""
}
reads_ch = Channel.fromFilePairs("/workspace/nextflow_tutorial/data/trimmed_fastq/SRR2584863_{1,2}.trim.fastq.gz")
workflow {
TUPLEINPUT(reads_ch)
}
Outputs
N E X T F L O W ~ version 21.04.3 Launching `process_tuple_input.nf` [curious_coulomb] - revision: 0bd8b0747e executor > local (1) [2b/b84295] process > TUPLEINPUT (1) [100%] 1 of 1 ✔ SRR2584863 SRR2584863_1.trim.fastq.gz SRR2584863_2.trim.fastq.gz
- In the same manner, an output channel emitting a tuple of values can be declared using the
tuplequalifier followed by the definition of each tuple element in the tuple. - In the code snippet below, the output channel would contain a tuple with the grouping key value as the Nextflow variable
sample_idand a list containing the files matching the following pattern"*.html"and"*.zip".
Create a new file process_fastqc.nf; add the following and run nextflow run process_fastqc.nf:
//process_fastqc.nf
nextflow.enable.dsl=2
process FASTQC {
input:
tuple val(sample_id), path(reads)
output:
tuple val(sample_id), path("*.html"), path("*.zip")
script:
"""
fastqc ${reads}
"""
}
reads_ch = Channel.fromFilePairs("/workspace/nextflow_tutorial/data/trimmed_fastq/SRR2584863_{1,2}.trim.fastq.gz", checkIfExists: true)
workflow {
FASTQC(reads_ch)
FASTQC.out.view()
}
The output is now a tuple containing the sample id and the .html and .zip files of the reads
Output
N E X T F L O W ~ version 21.04.3 Launching `process_fastqc.nf` [chaotic_waddington] - revision: c68f359bf9 executor > local (1) [3b/db0fae] process > FASTQC (1) [100%] 1 of 1 ✔ [SRR2584863, [nextflow_tutorial/channels/work/3b/db0fae4cf87ad545bf3666b626f55f/SRR2584863_1.trim_fastqc.html, nextflow_tutorial/channels/work/3b/db0fae4cf87ad545bf3666b626f55f/SRR2584863_2.trim_fastqc.html], [nextflow_tutorial/channels/work/3b/db0fae4cf87ad545bf3666b626f55f/SRR2584863_1.trim_fastqc.zip, nextflow_tutorial/channels/work/3b/db0fae4cf87ad545bf3666b626f55f/SRR2584863_2.trim_fastqc.zip]]
CLICK HERE for conditional execution directive of a process
- The `when` declaration allows you to define a condition that must be verified in order to execute the process. This can be any expression that evaluates a boolean value; `true` or `false`. - It is useful to enable/disable the process execution depending on the state of various inputs and parameters. In the example below the process `CONDITIONAL` will only execute when the value of the `chr` variable is less than or equal to 5:
//process_when.nf
nextflow.enable.dsl=2
process CONDITIONAL {
input:
val chr
when:
chr <= 5
script:
"""
echo ${chr}
"""
}
chr_ch = channel.of(1..22)
workflow {
CONDITIONAL(chr_ch)
}
Output
4 5 2 3 1
Directives
- Directive declarations allow the definition of optional settings, like the number of
cpusand the amount ofmemory, that affect the execution of the current process without affecting the task itself. - They must be entered at the top of the process body, before any other declaration blocks (i.e.
input,output, etc).
Note: You do not use
=when assigning a value to a directive.
- Directives are commonly used to define the amount of computing resources to be used or extra information for configuration or logging purposes.
For example, the process below uses three directives: tag, cpus, and label.
Modify process_index.nf as shown below and run nextflow run process_index.nf:
//process_index.nf
nextflow.enable.dsl=2
process BWA_INDEX {
tag {"BWA_INDEX $genome"}
label 'process_low'
cpus 1
input:
path genome
output:
path("*")
script:
"""
bwa index ${genome}
"""
}
ref_ch = Channel.fromPath("/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta")
workflow {
BWA_INDEX(ref_ch)
}
Output
N E X T F L O W ~ version 21.04.3 Launching `process_directive.nf` [nauseous_liskov] - revision: c8326ee33a executor > local (1) [0c/15e45d] process > BWA_INDEX (BWA_INDEX ecoli_rel606.fasta) [100%] 1 of 1 ✔
- The
tagdirective allows you to give a custom tag to each process execution. This tag makes it easier to identify a particular task (executed instance of a process) in a log file or in the execution report. - The second directive
labelallows the annotation of processes with mnemonic identifiers of your choice. Labels are useful to organize workflow processes in separate groups which can be referenced in the configuration file to select and configure subsets of processes having similar computing requirements. - The third directive
cpusallows you to define the number of CPUs required for each task.
A complete list of directives is available at this link.
Organising outputs - PublishDir directive
- Nextflow manages intermediate results from the pipelines expected outputs independently.
- Files created by a
processare stored in a task specific working directory which is considered as a temporary. Normally this is under theworkdirectory , that can be deleted upon completion. - The files you want the workflow to return as results need to be defined in the
processoutputblock and then the output directory specified using the directivepublishDir.
publishDir <directory>, parameter: value, parameter2: value ...
For example if we want to capture the results of the BWA_INDEX process in a results/bwa_index output directory we need to define the files in the output and specify the location of the results directory in the publishDir directive.
Modify process_index.nf as shown below and nextflow run process_index.nf:
//process_index.nf
nextflow.enable.dsl=2
process BWA_INDEX {
tag {"BWA_INDEX $genome"}
label 'process_low'
cpus 1
publishDir "results/bwa_index"
input:
path genome
output:
path("*")
script:
"""
bwa index ${genome}
"""
}
ref_ch = Channel.fromPath("/workspace/nextflow_tutorial/data/ref_genome/ecoli_rel606.fasta")
workflow {
BWA_INDEX(ref_ch)
}
We can use the UNIX command tree to examine the contents of the results directory; tree results :
results/ └── bwa_index ├── ecoli_rel606.fasta.amb -> nextflow_tutorial/processes/work/de/5b39abb456c91bd6e8436991be1de0/ecoli_rel606.fasta.amb ├── ecoli_rel606.fasta.ann -> nextflow_tutorial/processes/work/de/5b39abb456c91bd6e8436991be1de0/ecoli_rel606.fasta.ann ├── ecoli_rel606.fasta.bwt -> nextflow_tutorial/processes/work/de/5b39abb456c91bd6e8436991be1de0/ecoli_rel606.fasta.bwt ├── ecoli_rel606.fasta.pac -> nextflow_tutorial/processes/work/de/5b39abb456c91bd6e8436991be1de0/ecoli_rel606.fasta.pac └── ecoli_rel606.fasta.sa -> nextflow_tutorial/processes/work/de/5b39abb456c91bd6e8436991be1de0/ecoli_rel606.fasta.sa 1 directory, 5 files
In the above example, the publishDir "results/bwa_index", creates a symbolic link -> to the output files specified by the process BWA_INDEX to the directory path results/bwa_index.
publishDir - The publishDir output is relative to the path the pipeline run has been launched. Hence, it is a good practice to use implicit variables like
projectDirto specify publishDir value.
publishDir parameters
- The
publishDirdirective can take optional parameters, for example themodeparameter can take the value"copy"to specify that you wish to copy the file to output directory rather than just a symbolic link to the files in the working directory. - Since the working directory is generally deleted on completion of a pipeline, it is safest to use
mode: "copy"for results files. The default mode (symlink) is helpful for checking intermediate files which are not needed in the long term.
publishDir "results/bwa_index", mode: "copy"
Full list here.
Manage semantic sub-directories
- You can use more than one
publishDirto keep different outputs in separate directories. - To specify which files to put in which output directory, use the parameter
patternwith a glob pattern that selects which files to publish from the overall set of output files. - In the example below, we will create an output folder structure in the directory “results”, which contains a separate sub-directory for FastQC HTML files (
pattern: "*.html") and FastQC ZIP files (pattern: "*.zip"),results/fastqc_htmlandresults/fastqc_zip. Remember, we need to specify the files we want to copy as outputs.
Modify process_fastqc.nf as shown below and run nextflow run process_fastqc.nf:
//process_publishDir_semantic.nf
nextflow.enable.dsl=2
process FASTQC {
tag {"FASTQC $sample_id"}
label 'process_low'
cpus 2
publishDir "results/fastqc_html", pattern: "*.html", mode: 'copy'
publishDir "results/fastqc_zip", pattern: "*.zip", mode: 'copy'
input:
tuple val( sample_id ), path( reads )
output:
tuple val( sample_id ), path( "*.html" ), path( "*.zip" )
script:
"""
fastqc ${reads}
"""
}
reads_ch = Channel.fromFilePairs("/workspace/nextflow_tutorial/data/trimmed_fastq/SRR2584863_{1,2}.trim.fastq.gz", checkIfExists: true)
workflow {
FASTQC(reads_ch)
}
Output
N E X T F L O W ~ version 21.04.3 Launching `process_fastqc.nf` [nice_thompson] - revision: 547df8c76d executor > local (1) [a5/57a4fc] process > FASTQC (FASTQC SRR2584863) [100%] 1 of 1 ✔
We can now use the tree results command to examine the results directory.
Output
results/ ├── fastqc_html │ ├── SRR2584863_1.trim_fastqc.html │ └── SRR2584863_2.trim_fastqc.html └── fastqc_zip ├── SRR2584863_1.trim_fastqc.zip └── SRR2584863_2.trim_fastqc.zip 2 directories, 4 files
Nextflow Patterns - If you want to find out common structures of Nextflow process, the Nextflow Patterns page collects some recurrent implementation patterns used in Nextflow applications.
Quick Recap
- Outputs for a process are defined using the
outputblocks.- You can group
inputandoutputdata from a process using thetuplequalifier.- The execution of a process can be controlled using the
whendeclaration and conditional statements.- Files produced within a process and defined as output can be saved to a directory using the
publishDirdirective.