Reproducible, scalable, and shareable analysis workflows with Nextflow
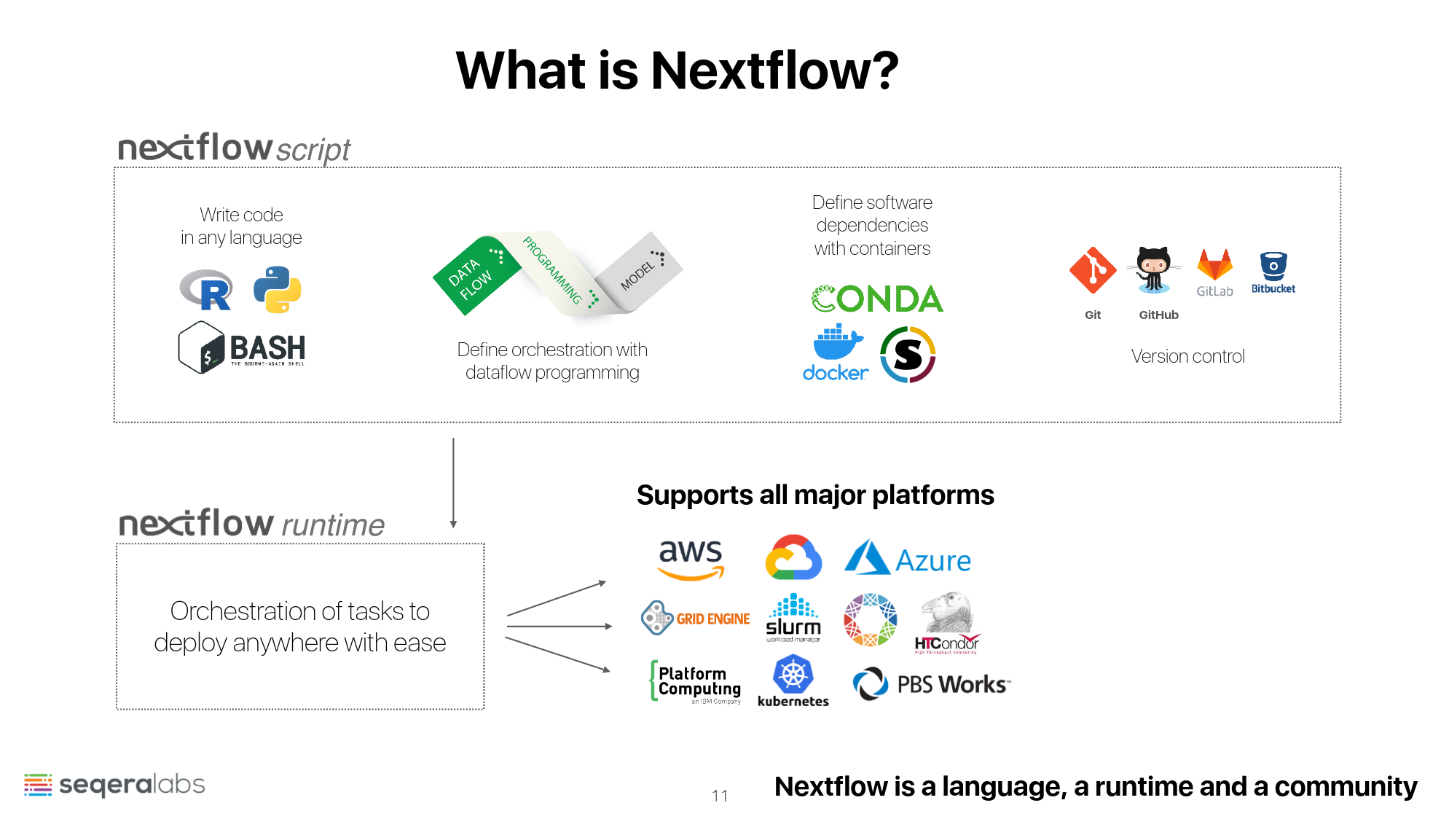
- Nextflow is a remarkably powerful and versatile workflow language designed for crafting scalable and reproducible scientific workflows.
- It adeptly integrates diverse software packages and environment management systems like Docker, Singularity, and Conda, enabling seamless coupling of existing pipelines written in popular scripting languages such as BASH, R, and Python.
- Nextflow streamlines the deployment and execution of workflows on cloud-based or high-performance computing (HPC) infrastructures, enhancing efficiency.
- Supported by nf-core, a community-driven initiative, Nextflow benefits from a curated collection of high-quality analysis pipelines.
Pre-Requisites
This lesson assumes a working understanding of the command line, familiarity with Conda, Docker/Singularity and running jobs on HPC cluster.
This lesson also assumes some familiarity with biological concepts, including the structure of DNA, nucleotide abbreviations, and the concept of genomic variation within a population.
Schedule & Learning Objectives

| Chapter | Learning Objectives |
|---|---|
| Session - 1 | 3 hours |
| 0. Setup | GitPod Link & Setup |
| How to install Nextflow? | |
| Data & Environment Setup | |
| 1. Nextflow Introduction | What is a workflow and what are workflow management systems? |
| Why should I use a workflow management system? | |
| What is Nextflow? | |
| What are the main features of Nextflow? | |
| What are the main components of a Nextflow script? | |
| How do I run a Nextflow script? | |
| How can I use the nextflow logs? | |
| 2. NF-Core | Where can I find existing bioinformatic pipelines? |
| What is nf-core tools? | |
| How do you run nf-core pipelines? | |
| How do you configure nf-core pipelines? | |
| How do you use nf-core pipelines offline? | |
| 3. NF-Core @ HPC | How do I configure nf-core pipelines to submit jobs to HPC cluster |
| Session - 2 | 3 hours |
| 4. Nextflow-Scripting | What language are Nextflow scripts written in? |
| How do I store values in a Nextflow script? | |
| How do I write comments Nextflow script? | |
| How can I store and retrieve multiple values? | |
| How are strings evaluated in Nextflow? | |
| How can I create simple re-useable code blocks? | |
| 5. Nextflow-Channels | How do I get data into Nextflow? |
| How do I handle different types of input, e.g. files and parameters? | |
| How do I create a Nextflow Channel? | |
| How can I use pattern matching to select input files? | |
| How do I change the way inputs are handled? | |
| 6. Nextflow-Processes | How do I run tasks/processes in Nextflow? |
| How do I pass parameters to a Nextflow script on the command line? | |
| How do I get data, files and values, into and out of processes? | |
| How do can I control when a process is executed? | |
| How do I control resources, such as number of CPUs and memory, available to processes? | |
| How do I save output/results from a process? | |
| 7. Nextflow-Workflow | How do I connect channels and processes to create a workflow? |
| How do I invoke a process inside a workflow? | |
| 8. Nextflow-Operators | How do I perform operations, such as filtering, on channels? |
| What are the different kinds of operations I can perform on channels? | |
| How do I combine operations? | |
| How can I use a CSV file to process data into a Channel? | |
| Session - 3 | 3 hours |
| 9. Simple Variant-Calling pipeline | How can I create a variant calling pipeline? |
| How do I print all the pipeline parameters by using a single command? | |
| How can I use conda with my pipeline? | |
| How do I know when my pipeline has finished? | |
| How do I see runtime metrics and execution information? | |
| 10. Nextflow configuration | What is the difference between the workflow implementation and the workflow configuration? |
| How do I configure a Nextflow workflow? | |
| How do I assign different resources to different processes? | |
| How do I separate and provide configuration for different computational systems? | |
| How do I change configuration settings from the default settings provided by the workflow? | |
| 11. Nextflow-Modules | How can I reuse a Nextflow process in different workflows? |
| How do I use parameters in a module? | |
| 12. Nextflow-Sub-workflows | How do I reuse a workflow as part of a larger workflow? |
| How do I run only a part of a workflow? | |
| 13. Nextflow-Reporting | How do I get information about my pipeline run? |
| How can I see what commands I ran? | |
| How can I create a report from my run? | |
| 14. Workflow caching and checkpointing | How can I restart a Nextflow workflow after an error? |
| How can I add new data to a workflow? | |
| Where can I find intermediate data and results? | |
| 15. nf-core/variantcall | Assembling the variant-calling workflow nf-core style |
| How can I add nf-core modules? | |
| 16. Contributing to nf-core/modules | How can i add a new module to nf-core? |
| Nextflow Useful Links |
Editors
Credits
Graeme R. Grimes, Evan Floden, Paolo Di Tommaso, Phil Ewels and Maxime Garcia Introduction to Workflows with Nextflow and nf-core. https://github.com/carpentries-incubator/workflows-nextflow 2021.
Josh Herr, Ming Tang, Lex Nederbragt, Fotis Psomopoulos (eds): “Data Carpentry: Wrangling Genomics Lesson.” Version 2017.11.0, November 2017, http://www.datacarpentry.org/wrangling-genomics/, doi: 10.5281/zenodo.1064254
- Happy Belly Bioinformatics (Jekyll site template and inspiration!)
Lee, (2019). Happy Belly Bioinformatics: an open-source resource dedicated to helping biologists utilize bioinformatics. Journal of Open Source Education, 4(41), 53, https://doi.org/10.21105/jose.00053